
La Unión Económica Euroasiática (EAEU) ha actualizado las normas de inspección farmacéutica mediante la Decisión n.º 60 (01/08/2025), introduciendo un control riguroso sobre las Buenas Prácticas de Laboratorio (BPL o GLP, Good Laboratory Practices). Este cambio normativo afecta directamente a los Sponsors (promotores) europeos, ya que las inspecciones ahora forman parte integral del proceso de registro de medicamentos en la región.
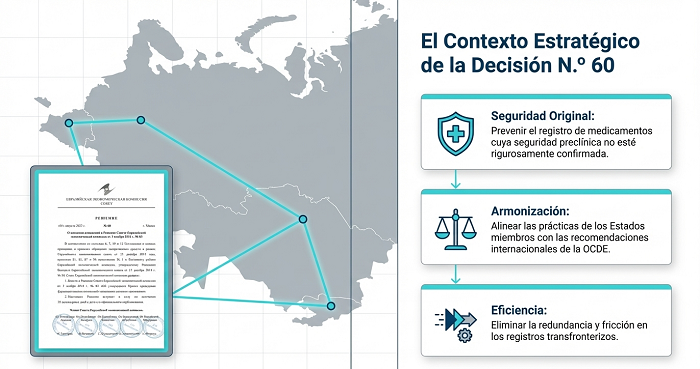
Un acceso único a cinco mercados
El beneficio principal de superar esta inspección es que otorga conformidad técnica para comercializar productos en los cinco estados miembros de la Unión: Armenia, Bielorrusia, Kazajistán, Kirguistán y Rusia. Un solo proceso de verificación abre la puerta a un mercado unificado, pero también traslada la responsabilidad de la calidad directamente al origen de los datos.
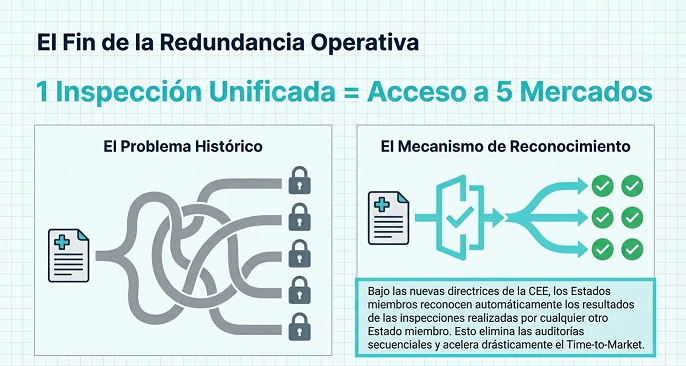
Inspecciones GLP en territorio de la UE y modalidad remota
Las normas establecidas en la Decisión n.º 60 son claras: las inspecciones no se limitan geográficamente a los países de la EAEU. Si los datos no clínicos del dossier se han generado en laboratorios situados en la Unión Europea (UE), estos centros son sujetos de inspección.
La normativa introduce además un enfoque basado en riesgos para determinar el formato de la auditoría:
- Inspecciones In-situ: Desplazamiento de los inspectores al laboratorio en la UE.
- Inspecciones Remotas: Uso de herramientas de audio y video en tiempo real para verificar instalaciones y registros de datos originales (raw data).
Cronograma detallado de la inspección GLP (Plazos críticos)
La Decisión n.º 60 impone tiempos estrictos que el Sponsor debe gestionar con precisión para evitar el colapso del proceso de registro.
Fase 1: Solicitud y Planificación
- Decisión de inspección GLP: La autoridad determina la necesidad de una inspección en un plazo de hasta 70 días hábiles tras recibir la solicitud de registro del medicamento.
- Notificación al Sponsor: Una vez decidida, la autoridad notifica al solicitante en un máximo de 5 días hábiles.
- Presentación de la solicitud de inspección GLP: El Sponsor tiene 15 días hábiles para presentar formalmente la solicitud ante el inspectorado farmacéutico tras recibir la notificación.
- Entrega de documentación técnica: Tras el requerimiento escrito de la inspección, el Sponsor debe entregar todos los documentos y registros necesarios para la planificación en un plazo de 30 días hábiles.
- Coordinación de fechas: El inspectorado acuerda las fechas definitivas en un plazo de 20 días hábiles tras recibir la solicitud formal.
Fase 2: Ejecución e Informes
- Preparación del programa: El inspector jefe debe finalizar el programa de inspección y los controles al menos 20 días hábiles antes de la fecha de inicio.
- Emisión del informe inicial: Tras finalizar la fase de auditoría, el inspector jefe redacta y envía el informe de inspección al laboratorio en un plazo máximo de 20 días hábiles. La clasificación de las deficiencias mira en la segunda parte de este artículo.
Fase 3: Respuesta y CAPA
- Plazo de respuesta del laboratorio: El centro inspeccionado (o el Sponsor) dispone de 20 días hábiles para enviar sus comentarios y, si se detectaron deficiencias mayores o críticas, el plan de Acciones Correctivas y Previvas (CAPA).
- Evaluación de la respuesta: La comisión inspectora tiene 15 días hábiles para evaluar si las medidas propuestas son aceptables y emitir el resumen final de evaluación.
- Informe final: El informe de inspección definitivo, firmado y con sus conclusiones, se envía al solicitante en un plazo de 3 días hábiles tras su firma.

El riesgo de la negativa: Un callejón sin salida para el registro
La Decisión n.º 60 es transparente en cuanto a los procedimientos: la inspección GLP se inicia como parte de los trámites de registro. Si un centro de investigación en la UE —ya sea por desconocimiento o por políticas internas— se niega a recibir a los inspectores de la EAEU o a facilitar el acceso a sus datos, las consecuencias para el Sponsor son críticas.
Una negativa de acceso conlleva la denegación automática del registro del medicamento. No existe la posibilidad de validar esos datos de forma alternativa si la autoridad ha decidido que la inspección es necesaria para confirmar la veracidad del dossier. Esto supone la pérdida de toda la inversión en el desarrollo del fármaco para ese mercado y un retraso estratégico incalculable.
El valor de la especialización: ¿Por qué contar con RuGMP?
Navegar por una inspección de la EAEU en territorio europeo requiere algo más que cumplimiento técnico; exige una gestión de riesgos bicultural y regulatoria. En RuGMP, entendemos que el éxito de estas inspecciones depende de dos pilares donde nuestra experiencia es fundamental:
- Consultoría y Auditoría Previa: Evaluamos si los laboratorios europeos y los acuerdos de calidad del Sponsor están alineados con las expectativas específicas de los inspectores de la EAEU antes de que estos lleguen al centro.
- Intérpretes Técnicos Especializados: Una inspección GLP se pierde o se gana en los matices. No basta con la traducción lingüística; es imprescindible contar con profesionales que comprendan la terminología científica y regulatoria para evitar que malentendidos lingüísticos se conviertan en «deficiencias críticas» de integridad de datos.
Conclusión
La Decisión n.º 60 cierra el círculo de la supervisión de calidad en la EAEU, extendiéndola a la fase preclínica. Los laboratorios en la UE deben ser conscientes de que sus datos son ahora el puente —o la barrera— para el acceso al mercado. La preparación proactiva y el soporte experto son las únicas herramientas para garantizar que la ciencia europea sea validada con éxito bajo este nuevo marco normativo.




EAEU GLP Inspections: Clasificación de Riesgos - ruGMP
[…] del dato desde su origen. Para los laboratorios y Sponsors europeos, lo más relevante es el enfoque basado en riesgos que determina cómo y qué será […]